| MitImpact id |
MI.5176 |
MI.5175 |
| Chr |
chrM |
chrM |
| Start |
7587 |
7587 |
| Ref |
T |
T |
| Alt |
C |
A |
| Gene symbol |
MT-CO2 |
MT-CO2 |
| Extended annotation |
mitochondrially encoded cytochrome c oxidase II |
mitochondrially encoded cytochrome c oxidase II |
| Gene position |
2 |
2 |
| Gene start |
7586 |
7586 |
| Gene end |
8269 |
8269 |
| Gene strand |
+ |
+ |
| Codon substitution |
ATG/ACG |
ATG/AAG |
| AA position |
1 |
1 |
| AA ref |
M |
M |
| AA alt |
T |
K |
| Functional effect general |
start_lost |
start_lost |
| Functional effect detailed |
start_lost |
start_lost |
| OMIM id |
516040 |
516040 |
| HGVS |
NC_012920.1:g.7587T>C |
NC_012920.1:g.7587T>A |
| HGNC id |
7421 |
7421 |
| Respiratory Chain complex |
IV |
IV |
| Ensembl gene id |
ENSG00000198712 |
ENSG00000198712 |
| Ensembl transcript id |
ENST00000361739 |
ENST00000361739 |
| Ensembl protein id |
ENSP00000354876 |
ENSP00000354876 |
| Uniprot id |
P00403 |
P00403 |
| Uniprot name |
COX2_HUMAN |
COX2_HUMAN |
| Ncbi gene id |
4513 |
4513 |
| Ncbi protein id |
YP_003024029.1 |
YP_003024029.1 |
| PhyloP 100V |
7.73 |
7.73 |
| PhyloP 470Way |
0.666 |
0.666 |
| PhastCons 100V |
1 |
1 |
| PhastCons 470Way |
0.14 |
0.14 |
| PolyPhen2 |
probably_damaging |
probably_damaging |
| PolyPhen2 score |
0.97 |
0.97 |
| SIFT |
deleterious |
deleterious |
| SIFT score |
0.0 |
0.0 |
| SIFT4G |
Damaging |
Damaging |
| SIFT4G score |
0.0 |
0.0 |
| VEST |
Neutral |
Neutral |
| VEST pvalue |
0.37 |
0.3 |
| VEST FDR |
0.5 |
0.45 |
| Mitoclass.1 |
damaging |
damaging |
| SNPDryad |
Pathogenic |
Pathogenic |
| SNPDryad score |
0.92 |
1.0 |
| MutationTaster |
Disease automatic |
Disease |
| MutationTaster score |
1 |
1 |
| MutationTaster converted rankscore |
0.81001 |
0.81001 |
| MutationTaster model |
complex_aae |
complex_aae |
| MutationTaster AAE |
A2Q |
A2Q |
| fathmm |
Tolerated |
Tolerated |
| fathmm score |
1.24 |
1.27 |
| fathmm converted rankscore |
0.36691 |
0.36146 |
| AlphaMissense |
. |
. |
| AlphaMissense score |
. |
. |
| CADD |
Deleterious |
Deleterious |
| CADD score |
2.728718 |
3.800415 |
| CADD phred |
21.0 |
23.4 |
| PROVEAN |
Damaging |
Damaging |
| PROVEAN score |
-5.73 |
-5.73 |
| MutationAssessor |
. |
. |
| MutationAssessor score |
. |
. |
| EFIN SP |
Damaging |
Damaging |
| EFIN SP score |
0.156 |
0.148 |
| EFIN HD |
Damaging |
Damaging |
| EFIN HD score |
0.016 |
0.014 |
| MLC |
Deleterious |
Deleterious |
| MLC score |
0.7 |
0.7 |
| PANTHER score |
. |
. |
| PhD-SNP score |
. |
. |
| APOGEE1 |
Pathogenic |
Pathogenic |
| APOGEE1 score |
0.79 |
0.83 |
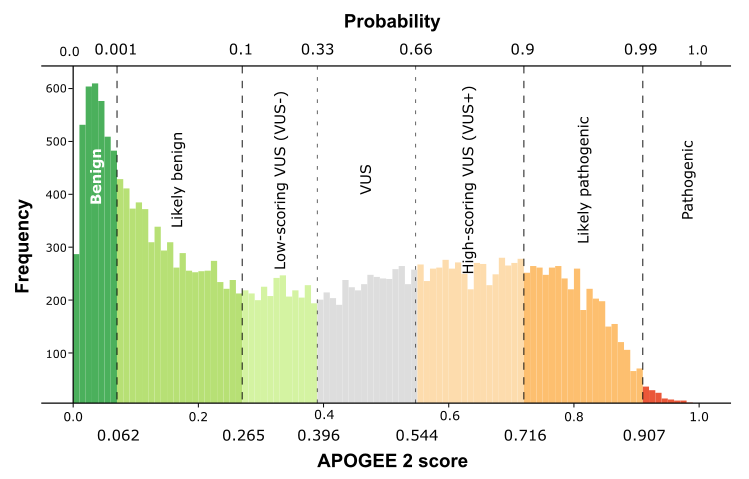
| APOGEE2 |
Likely-pathogenic |
Likely-pathogenic |
| APOGEE2 score |
0.875036505601518 |
0.804333931513274 |
| CAROL |
deleterious |
deleterious |
| CAROL score |
1.0 |
1.0 |
| Condel |
neutral |
neutral |
| Condel score |
0.02 |
0.02 |
| COVEC WMV |
deleterious |
deleterious |
| COVEC WMV score |
4 |
4 |
| MtoolBox |
deleterious |
deleterious |
| MtoolBox DS |
0.79 |
0.79 |
| DEOGEN2 |
Tolerated |
Tolerated |
| DEOGEN2 score |
0.231232 |
0.129293 |
| DEOGEN2 converted rankscore |
0.59750 |
0.45748 |
| Meta-SNP |
. |
. |
| Meta-SNP score |
. |
. |
| PolyPhen2 transf |
. |
. |
| PolyPhen2 transf score |
. |
. |
| SIFT_transf |
. |
. |
| SIFT transf score |
. |
. |
| MutationAssessor transf |
. |
. |
| MutationAssessor transf score |
. |
. |
| CHASM |
Neutral |
Neutral |
| CHASM pvalue |
0.22 |
0.36 |
| CHASM FDR |
0.8 |
0.8 |
| ClinVar id |
9658.0 |
. |
| ClinVar Allele id |
24697.0 |
. |
| ClinVar CLNDISDB |
MONDO:MONDO:0009068,MedGen:C5435656,OMIM:220110,Orphanet:254905 |
. |
| ClinVar CLNDN |
Cytochrome-c_oxidase_deficiency_disease |
. |
| ClinVar CLNSIG |
Pathogenic |
. |
| MITOMAP Disease Clinical info |
Mitochondrial Encephalomyopathy |
. |
| MITOMAP Disease Status |
Cfrm [LP] |
. |
| MITOMAP Disease Hom/Het |
-/+ |
./. |
| MITOMAP General GenBank Freq |
0.0% |
. |
| MITOMAP General GenBank Seqs |
0 |
. |
| MITOMAP General Curated refs |
21457906;10205264 |
. |
| MITOMAP Variant Class |
disease |
. |
| gnomAD 3.1 AN |
56432.0 |
. |
| gnomAD 3.1 AC Homo |
0.0 |
. |
| gnomAD 3.1 AF Hom |
0.0 |
. |
| gnomAD 3.1 AC Het |
0.0 |
. |
| gnomAD 3.1 AF Het |
0.0 |
. |
| gnomAD 3.1 filter |
npg |
. |
| HelixMTdb AC Hom |
. |
. |
| HelixMTdb AF Hom |
. |
. |
| HelixMTdb AC Het |
. |
. |
| HelixMTdb AF Het |
. |
. |
| HelixMTdb mean ARF |
. |
. |
| HelixMTdb max ARF |
. |
. |
| ToMMo 54KJPN AC |
. |
. |
| ToMMo 54KJPN AF |
. |
. |
| ToMMo 54KJPN AN |
. |
. |
| COSMIC 90 |
. |
. |
| dbSNP 156 id |
rs199474825 |
. |